Some times it is difficult to completely get rid of the protonated solvent in your sample, which results in huge peaks that could overwhelm your solute signals. In such cases you can perform a solvent suppression technique. Many solvent suppression techniques are available. An easy and effective technique is called WET, which also has the ability to suppress multiple solvent peaks. Follow these steps to set it up:
- Lock and shim your sample. Good shimming will assure optimal solvent suppression. Collect a proton spectrum, which you will use to define the peak regions that you wish to suppress.
- With the proton spectrum on the screen, click Acquire in the main menu, Click Options, then select Setup Selective 1D Expts:

3. Click 1D Selective Experiment Setup. You will see an instruction of the expt setup. Read it to get a basic idea. Then click Close.

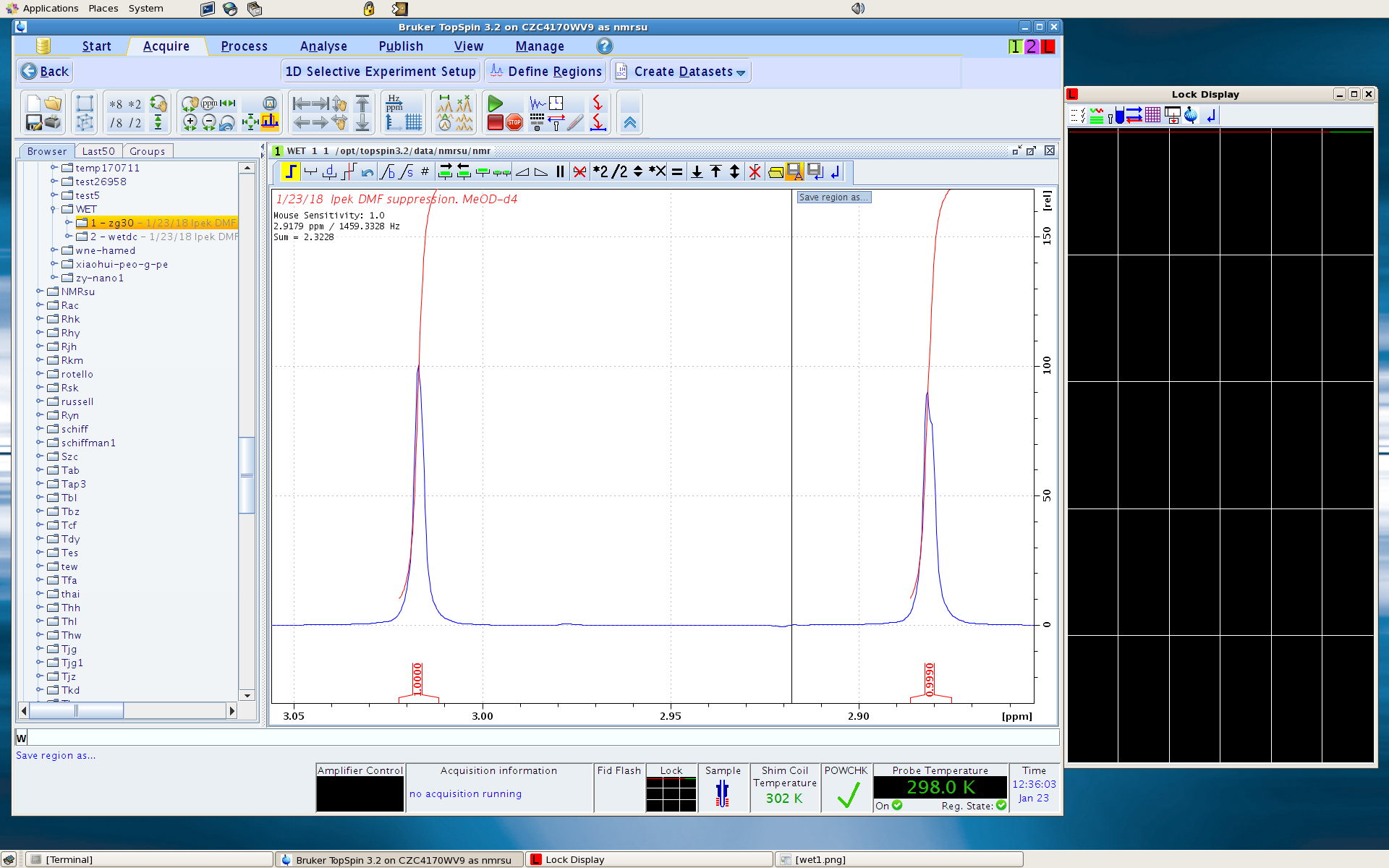
4. Click Define Regions. You are given the integration module for this task, though your job here is not integrating the peak areas, but rather to select the peak regions that you wish to suppress. I suggest that you select a narrow region around each solvent peak which covers most of the peak intensity, like shown below. Don’t worry too much about the tails. If you select a wide region, you will suppress solute peaks in the region. It is always a good idea to be a little conservative in the beginning. When done, click Save Region as…, and select “Save regions to ‘reg’“. Then click Save and Return.
5. Click Create Datasets, then select “Mult. Solvent Suppr./WET”

6. It will ask you NS and first EXPNO (experiment number). Usually NS of 16 is good enough. For first EXPNO, give an experiment number that you have not used (let’s say 3).
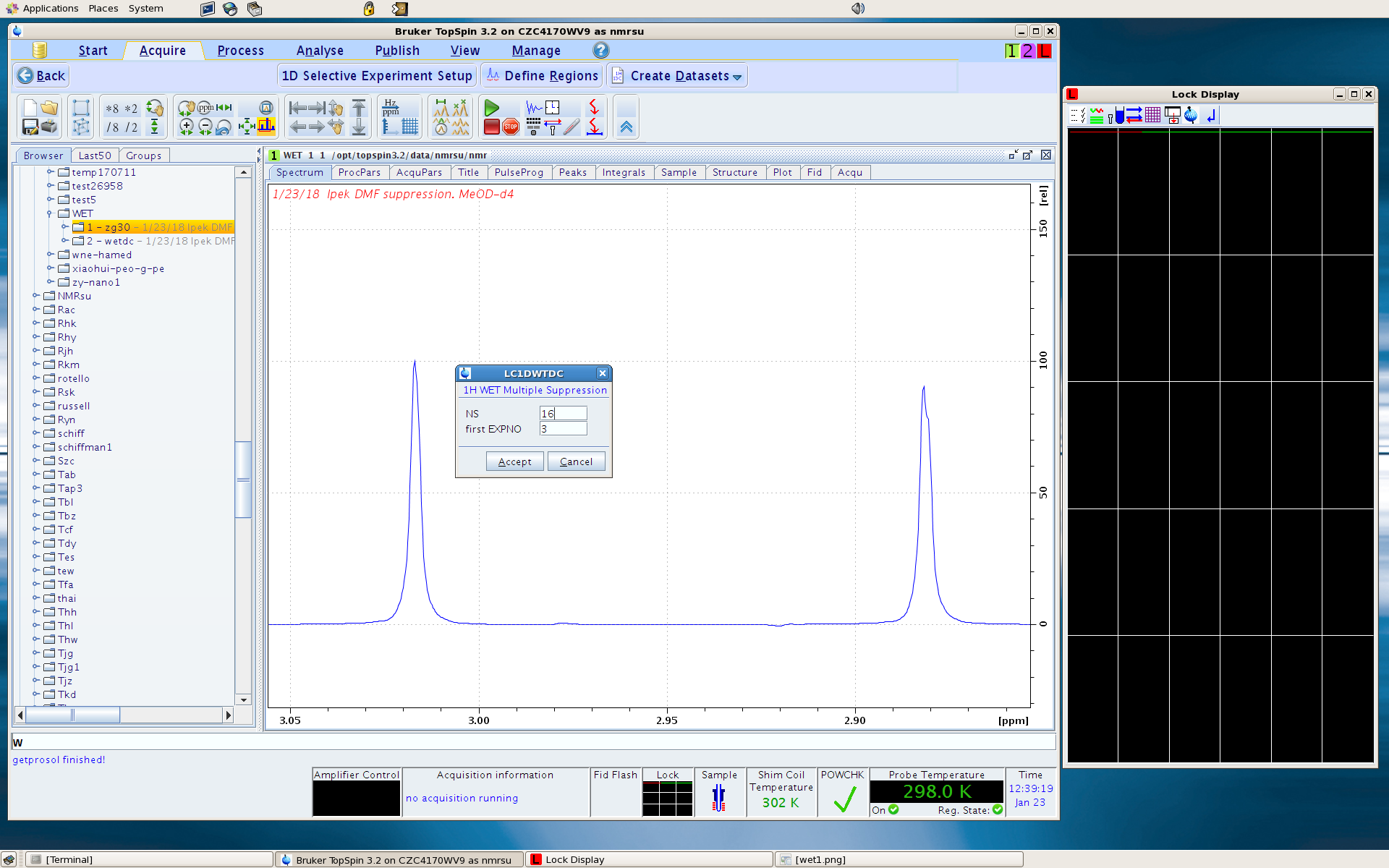
7. A window shows up summarizing the peak position that you have selected for suppression. Click Cancel – you will need to do atma (to tune 13C, as the WET uses 13C decoupling to remove the 13C satellites of the solvent peaks) and customize some parameters before you run the expt.

8. Issue command “re 3“, which will read in experiment #3, which you just created. Perform atma which will tune both 13C and 1H. Then type d1 and change it to 6 or 10. Since the WET experiment uses 90 deg excitation, T1 relaxation for many solutes might not be complete for the default d1 of 3 s, so it is safer to use a longer d1.
9. Do rga, then zg.
In the following picture, the blue spectrum is a proton spectrum of a sample with protonated DMF. The red spectrum is a WET spectrum which have the three DMF signals suppressed.








